Nava Whiteford1, Andrew Heron2, Leonard McCline3, Ales Flidr3, Jacob Swett4, Ajay Karpur5
1 Whiteford Research
2 Independent
3 Convergent Research
4 Blueprint Biosecurity
5 Open Philanthropy
Cite as: Whiteford, N., Heron, A., McCline, L., Flidr, A., Swett, J., & Karpur, A.. Towards Ubiquitous Metagenomic Sequencing: A Technology Roadmap. https://doi.org/10.17605/OSF.IO/8JU9M
Project was funded by Open Philanthropy and coordinated by Convergent Research.
Supplementary material: MGS sensitivity literature review, Limit of detection model, Sequencer comparison model
Metagenomic sequencing (MGS) has shown promise for infectious disease diagnostics and pandemic preparedness but has not yet reached widespread clinical adoption due to limitations such as high costs and complex workflows. This technology roadmap proposes target specifications for a MGS diagnostic device to enable routine use: sensitivity comparable to polymerase chain reaction, time-to-answer under 1 hour, cost per test under $10, and a portable, affordable instrument. We estimate that throughput of 1-10 million reads per hour with modest read lengths >25 base pairs and accuracy >95% could robustly detect most pathogens in human respiratory samples. Existing sequencing platforms do not meet this combination of targets, so focused technology development is needed. Nanopore and single-molecule optical sequencing are highlighted as promising approaches if optimized for the proposed specifications rather than long reads and maximum accuracy. Realizing ubiquitous MGS may require push and pull incentives for innovation. A low-cost, rapid MGS diagnostic appears technically feasible and could greatly enhance pandemic preparedness.
The COVID-19 pandemic has revealed the limitations of our ability to identify emerging pathogens. SARS-CoV-2 circulated in human populations as an unknown pneumonia, undetected by standard-of-care diagnostics, for an estimated 4-8 weeks [1] before a novel coronavirus was identified in infected samples by unbiased MGS [2]. This delay lost valuable time for addressing the pandemic, which has resulted in millions of deaths and trillions of dollars in economic costs [3].
Imagine if, instead, every clinician and patient around the world had access to a simple device capable of detecting any virus, bacterium or other pathogen causing disease in a symptomatic patient. Such a world would be much better positioned to diagnose and treat infectious disease and to detect novel emerging pathogens before they cause a devastating pandemic.
MGS has demonstrated the potential to become such a near-universal diagnostic [4-7] and is already saving lives by informing clinical decisions for a variety of symptoms and sample types [8]. However, it is not yet ready for prime time: complex workflows and high costs prevent widespread adoption [4]. As a result, MGS has had a limited impact on the management of the pandemic at the point of care. Therefore, regardless of their value for public health, sequencing-based assays have to become comparable with the standard of care to be considered a viable alternative.
While isothermal amplification methods and other new modalities gained in use during COVID-19, assays based on the qPCR were by far the most widely used molecular diagnostic. Currently, qPCR is considered the gold standard for the detection of respiratory pathogens, offering high sensitivity, specificity, and a rapid turnaround time [9]. In this paper, we use qPCR as a benchmark and outline the specifications for a clinical MGS device for viral respiratory infection diagnostics. Having outlined these specifications, we discuss the most promising candidate technologies that may meet these requirements.
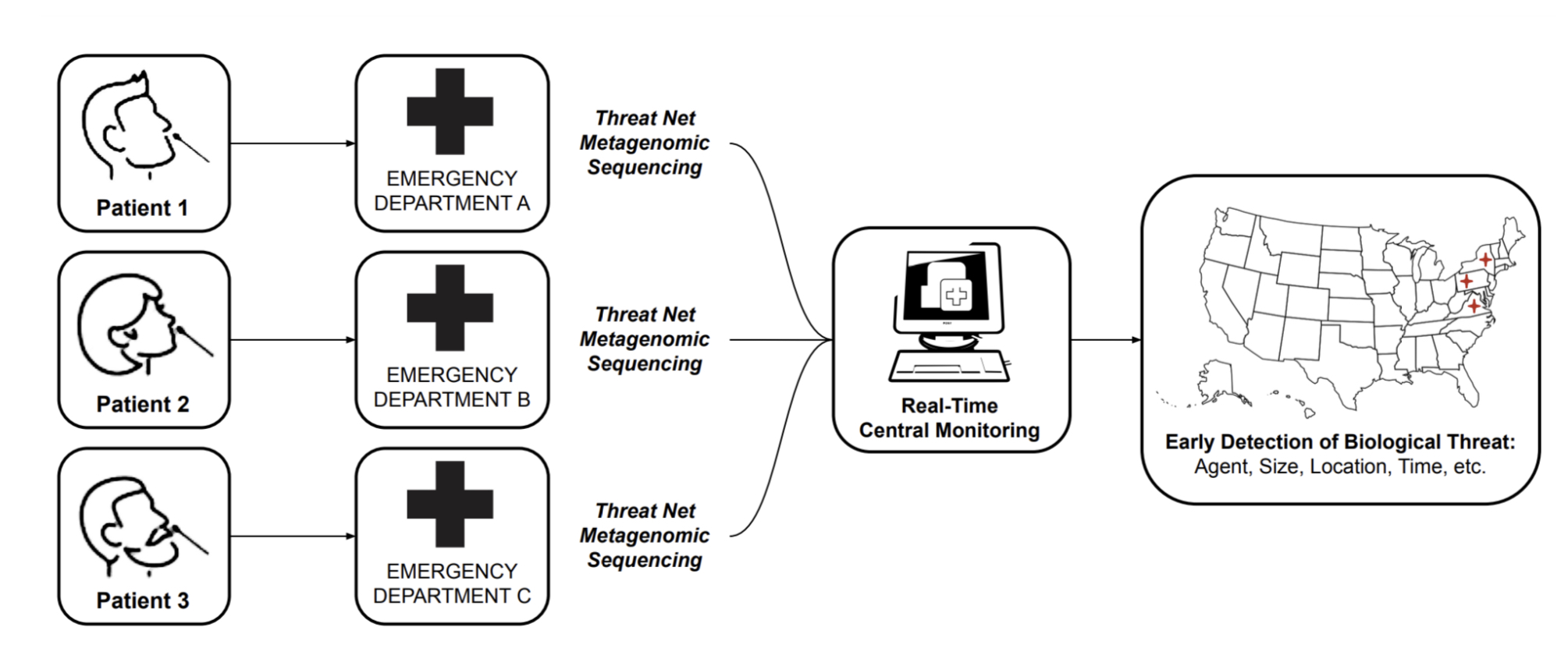
MGS can be leveraged for early detection in a number of ways, including sequencing sites searching for emerging pathogens in wastewater and waterways [11], or strategic testing of "sentinel" populations. While these approaches should certainly form an important part of a layered early detection system, we focus this roadmap report solely on the vision of MGS deployed widely at the point of care for detection of pathogens in human respiratory samples. The key reasons for this focus are:
Distinguishing between signal and noise is likely to be easier in human respiratory samples than in environmental ones.
Early detection of pathogens will only be enabled when MGS is deployed at a relatively large scale [10]. If MGS proves to be clinically useful and cost-effective, it can scale up naturally within the current health-economic system [12], without the need for continued public or philanthropic support above those of current diagnostics.
Detecting a pathogen of concern in an individual sample immediately enables effective quarantining and contact tracing.
Widely available MGS testing would put humanity in a position where mass testing of a novel pathogen is possible from "day zero" of a potential pandemic, saving the months it would take to develop and approve novel primers for PCR tests.
With the ongoing COVID-19 pandemic, we have seen that sequencing has had a limited impact relative to its huge potential: while it has aided initial sequence identification and variant tracking, a number of bottlenecks prevent its widespread adoption directly at the point of care. In this report, we analyze these bottlenecks and ask what it would take to make the technology for MGS truly ubiquitous, fit for developed and low-income countries alike in a 10-year time frame.
What is it going to take to make MGS ubiquitous in clinics around the world? A necessary condition is the existence of an affordable, easy-to-use technical solution.
A natural success story to draw on is that of point-of-care polymerase chain reactions (PCR) machines. Initially, these devices were bulky, expensive, and limited to specialized laboratories, requiring hours to produce results. Over time, they have been transformed into affordable, user-friendly devices that can quickly deliver results with the "push of a button", eliminating the need for specialized personnel. According to a WHO report [9], real-time PCR devices have led to wider adoption of molecular diagnostics due to their improved rapidity, sensitivity, reproducibility and the reduced risk of carry-over contamination. With the exception of sensitivity, all of these problems currently still plague MGS.
A concrete example of this success is the Cepheid GeneXpert [14] device. Originally developed for the detection of anthrax, it has been adapted to many other infectious diseases following collaboration between Cepheid and international organizations and philanthropic bodies. Through successive rounds of cost-optimization, the device arrived at a point where it became practical even in developing countries for testing infections such as tuberculosis or HIV. This resulted in an overall installed base of some 22,000 devices even prior to the COVID pandemic [15], which increased to 40,000 between 2019 and 2022 [16]. This enabled relatively rapid development of primers for testing the presence of SARS-CoV-2 in clinical samples without the need for developing novel infrastructure.
PCR devices can also test for multiple pathogens or genes (such as those conferring antimicrobial resistance) in parallel. The BioFire FilmArray [17], for instance, offers simultaneous sensitive detection of 20-40 targets in samples including respiratory (sputum, bronchoalveolar lavage), blood culture, cerebrospinal fluid or gastrointestinal.
However, PCR cannot in principle detect unknown or changing targets. For example, a novel gene conferring drug resistance, or a novel virus strain, require the design of novel primers. Perhaps more importantly, the emergence of a completely novel pathogen (in recent decades, consider, SARS-CoV-1 and 2, HIV, Ebola and Marburg virus, for instance) would go completely undetected.
In principle, then, MGS has a clear advantage, as it can detect any pathogen, whether bacterial, viral, fungal or otherwise. Despite this advantage, however, it is difficult to imagine that MGS might become truly ubiquitous without being close to matching PCR on the set of characteristics that made it successful.
In particular, we believe that MGS-based diagnostics should aspire to meet these requirements:
Sensitivity comparable to qPCR (ideally matching a Ct of 35).
Workflow should be “push-button”, requiring minimal operator time and skill (<5 minutes).
Time to answer should be less than 1 hour.
The cost of a single test (COGS) should be comparable to qPCR ($10).
The device itself should be affordable (ideally < $10,000) and compact, ideally portable.
Supply chains should not be overly complex.
As Table 1 (supplementary note) illustrates, no sequencing device currently comes close to meeting these specifications. Sensitivity itself is already achievable with sufficient sequencing depth (see section on platform requirements), but incompatible with the other requirements of cost and time-to-answer. Workflows involve complex sequential steps in both sample and library preparation and typically require hours of work by trained experts. Automated and integrated solutions such as VolTRAX for nanopore sequencing [31] or NeoPrep for Illumina [32] are only available at high costs for specialized laboratories. Cost per sample can be reduced below $10 only by sequencing many samples in parallel, which is not practical in the point-of-care context, as this step introduces a significant delay in time-to-answer.
| Platform | Method | Fastest lib. prep | Fastest run time | RNA? | COGS | Ref |
|---|---|---|---|---|---|---|
| Illumina NovaSeq 6000 | SBS | 1.5 hr | 13 hr | No | $50 | [18] |
| Illumina MiSeq | SBS | 1.5 hr | 1.5 hr | No | $30 | [19] |
| Illumina iSeq | SBS | 1.5 hr | 2 hr | No | $50 | [20] |
| Ion Torrent GeneStudio S5 | Ion Semi. SBS | 2 hr | 10 min | No | $50 | [21]–[24] |
| ONT MinION | SM Nanopore | 10 min | <1 hr | Yes | $250 | [25]–[28] |
| SeqLL (Helicos) | SMO SBS | 0 min | 7 d | Yes | $50 | [29] |
| Pacific Biosciences Sequel II | SMO SBS | 3 hr | <1 hr | No | $50 | [30] |
The need for rapid results speaks against the majority of sequencing platforms based on colony-based approaches. The cluster generation step alone takes at least 60 minutes [33]. In addition, this approach requires a large set of reagents that would add significant complexity and cost to the design of a sample-to-answer system.
Although the emergence of new companies (e.g. MGI [34], Singular Genomics [35]) is likely to decrease consumable and device costs by driving down margins and enabling innovations, the new players are unlikely to change this fundamental limitation. To our knowledge, the most serious attempt at decreasing the time requirements is the use of Lighting Terminators [36].
While this and other future innovations could make some colony-based approaches viable, a more natural category to focus on is that of single-molecule approaches. Single-molecule approaches have the advantage of potentially minimal library preparation and compatibility with a real-time readout, with results delivered in minutes following sample and library preparation. The two main approaches in this category as of 2023 are nanopore sequencing and single-molecule optical (SMO) sequencing.
As of 2023, the cheapest available instrument for runs on single samples is ONT’s Flongle, whose consumables sell for $90 [37] and likely costs around $50 to make [38]. Optimized clinical workflows utilizing real-time sequencing with ONT’s MinION can achieve a time-to-answer of 6 hours [7]. This is driven primarily by sample and library preparation, as sequencing itself takes less than 1 hour. Even workflows optimized for the ICU setting require a clinical microbiology laboratory to execute.
In the rest of this report, we ask what requirements a sequencing device has to meet in order to match all of the above criteria. In particular, we ask:
How could the sample and library preparation workflows be automated to meet the cost and time-to-answer requirements?
How does clinical sensitivity translate into sequencing device specifications?
What does this imply for sequencing technology development goals and what steps might be necessary to direct and accelerate this development?
Perhaps the greatest contrast between sequencing and PCR tests today lies in the complexity of workflows. As previously mentioned, even workflows optimized for the ICU setting require a clinical microbiology laboratory to execute [8]. Interviews with practitioners at the forefront of clinical MGS adoption reveal that training new personnel in MGS workflows takes months and results vary significantly based on operator skill.
In contrast, PCR assays are straightforward. To obtain a clinical answer with the previously mentioned Cepheid GeneXpert, the user places the sample into a cartridge, inserts it into the device, and waits for approximately 45 minutes to obtain the result. This simplicity is currently unattainable in the sequencing world, where the standard procedure includes complex sample and library preparation workflows. Given the ease of use and efficacy of Cepheid’s sample-to-answer RT-PCR platform, it is valuable to explore whether similar principles could be applied to sequencing technologies. Can we envision a device akin to Cepheid’s, but centered around sequencing instead of qPCR? This would potentially offer streamlined, rapid sequencing workflows, which could dramatically accelerate the field.
Cepheid’s first and second generation instruments were incapable of processing raw samples directly. To establish a comprehensive sample-to-answer system, Cepheid tackled this limitation by incorporating sample preparation into the platform. This was accomplished by designing a fluidic cartridge capable of processing raw samples and concurrently integrating a qPCR reaction tube. Reagents can be preloaded, with no fluidic coupling to the instrument. The instrument interfaces with the cartridge via a reagent selection valve and plunger. The cost of goods for the cartridge, including reagents, has been estimated at $10 [39] and marketed at prices typically exceeding $20.
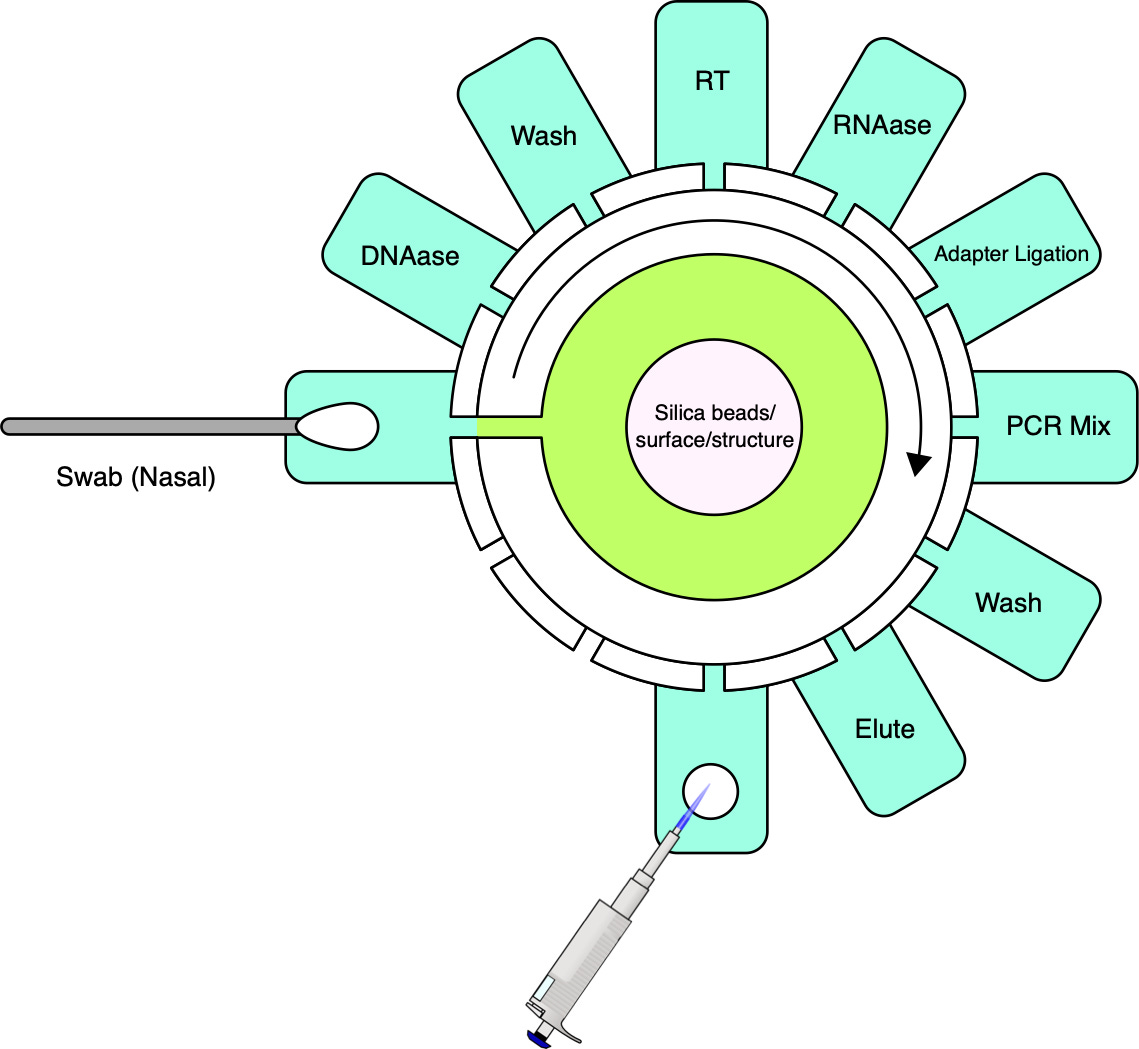
.
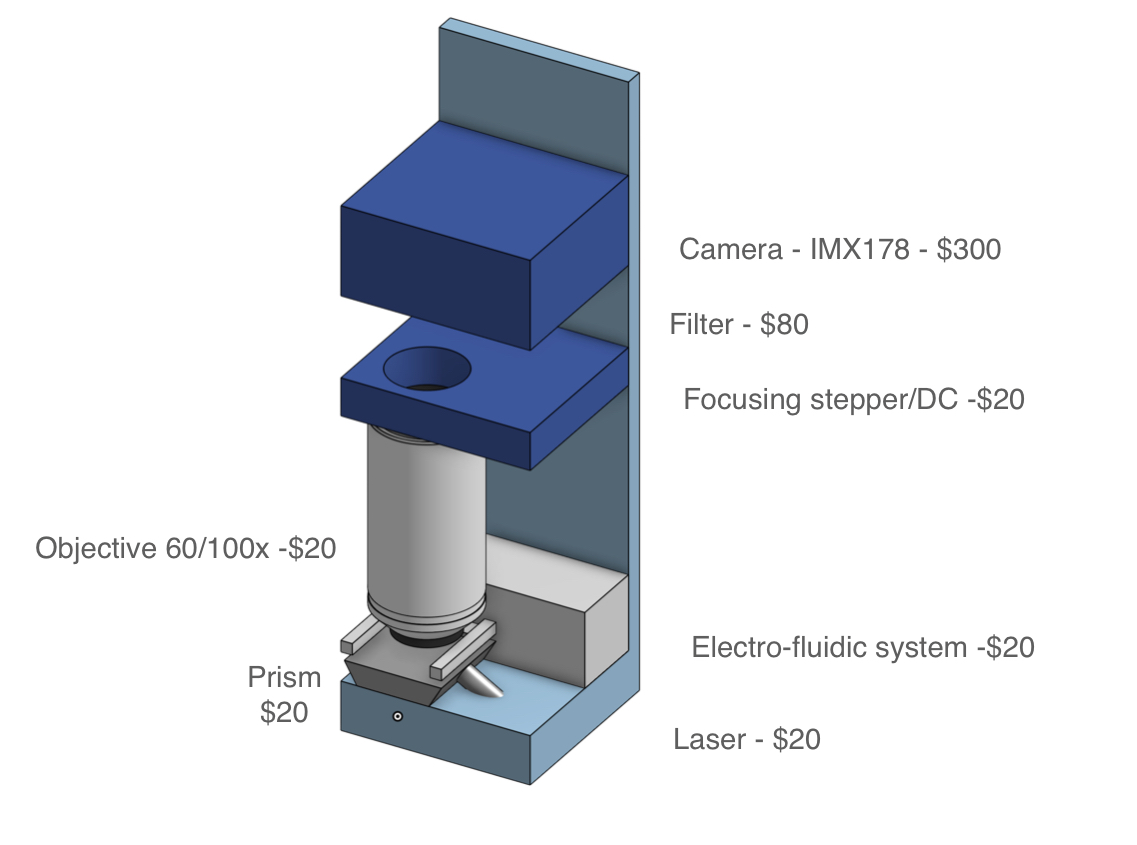
While achieving the same simplicity for MGS may seem like a tall order, it is important to note that currently available cartridges have been designed to be versatile and serve a number of applications. For MGS applications, workflows can be optimized for specific sample types such as nasopharyngeal, upper nasal, or saliva, along with a consistent input volume and exclusive RNA sequencing. If sequencing is stripped down to its bare essentials (see cartridge sketch figure), the complexity need not be much greater than that of qPCR.
Like qPCR, sample preparation will, for the foreseeable future, need to involve cell lysis, nucleic acid extraction and, unless direct RNA sequencing becomes more reliable, a reverse transcription step to convert RNA to complementary DNA (cDNA). Further needed steps include the removal of unwanted nucleic acid material, in this case, digesting DNA using DNAase, and, in most cases, the addition of adapters for the sequencing platform in question. For platforms with relatively high input requirements such as ONT, random amplification may be necessary to reach a lower threshold of nucleic acids in a sample without introducing undue bias [41].
Developing a cartridge for this use case and integrating the whole system into one box can be done with relatively little technical risk. Why, then, has no one developed such a system? The key reason, we believe, is the lack of a platform that could achieve the sufficient sequencing depth at a low enough cost without the need to analyze multiple samples in parallel. In the next section, we ask what the required sequencing depth for an MGS diagnostic is likely to be.
The most important question any candidate test for infectious disease has to address is whether its sensitivity of detection is sufficient. While PCR and sequencing will offer a different profile of costs and benefits and need not compete for the same niche, the performance of PCR offers a good starting point for thinking about sensitivity requirements for MGS. In the following discussion, we focus on viral pathogens in respiratory samples, as their relative abundances is generally much lower than those of bacteria and viruses are therefore likely to drive the requirements for sequencing depth.
It is worth emphasizing that MGS and PCR yield results that differ in kind. PCR delivers binary information about the presence or absence of a pathogen, and indirectly about its abundance in the sample. In contrast, a MGS run will identify a number of nucleic acid fragments belonging to a pathogen of interest, in addition to fragments of the host and the non-pathogenic microbiome. Hence, an additional judgment call is necessary for determining whether a given result should be taken as indication of infection [4,6].
In clinical practice, cutoffs for clinical significance will likely be determined for each pathogen or pathogen class as information about typical abundance in non-infected individuals is accumulated. As an example, in a pulmonary sample study, Zinter et al. used two criteria: a normalized score of reads per million (rpm) and the deviation in abundance from other samples in the cohort and determined the cutoff for bacteria as a deviation of Z ≥ 2 or 10 rpm, and for viruses and fungi as 1 rpm [6]. Miller et al. developed threshold criteria based on the detection of non-overlapping reads from ≥ 3 distinct genomic regions [42].
With these goals in mind, what sequencing depth is required? PCR tests are characterized by a high sensitivity, or very low limit of detection (LoD): in principle, they can detect the presence of a target fragment with only a handful of copies present in a sample. Sequencing a human clinical sample can obviously achieve very high levels of sensitivity: a sequencing run of terabases (Tb) on a single sample should readily detect even pathogens that are very low in abundance. At a depth of 40M reads per sample, MGS was shown to consistently achieve a sensitivity on par with if not greater than PCR [40,43]. However, when the requirements of cost and time-to-answer are added, practical sensitivity of sequencing has to be determined.
PCR has a key advantage over sequencing: because PCR targets a short, unique region of a genetic sequence, it is insensitive to background material. A high fraction of human material (mostly rRNA in the case of RT-qPCR) or bacterial material will have only a minor effect on the sensitivity of qPCR. In other words, the limit of detection relies on the sample’s absolute abundance of the target. MGS, however, is sensitive to background material. To ensure that the target of interest is detected, one must also sequence through background fragments until the target is reached. Thus, the sensitivity of MGS relies on the relative abundance of the target among the other nucleic acids in a sample.
In typical human clinical samples, host nucleic acids are orders of magnitude more abundant than those of the pathogen. For example, the typical fraction of SARS-CoV-2 RNA in nasopharyngeal samples was between 0.01% (or one fragment in 10,000) and 0.001% (one in 100,000). However, viral load has been found to span some 8 orders of magnitude, with loads as low as tens of copies/mL found in more than 1% of cases [44].
| Target Fraction | Target in Sample | Target Reads (10M) | Target Reads (1M) | Target Reads (1M, rRNA dep.) | Pred. Ct | |
|---|---|---|---|---|---|---|
| 1 × 10−7 | 1.8 × 103 | 1 | 0.1 | 0.2 | 39.1 | |
| 1 × 10−6 | 1.8 × 104 | 10 | 1 | 2 | 35.8 | |
| 1 × 10−5 | 1.8 × 105 | 100 | 10 | 25 | 32.4 | |
| 1 × 10−4 | 1.8 × 106 | 1000 | 100 | 250 | 29.1 | |
| 1 × 10−3 | 1.8 × 107 | 10000 | 1000 | 2496 | 25.7 |
The expected number of reads from the pathogen of interest is a straightforward function of the relative fraction of the pathogen in the sample, and the number of fragments sequenced in a run. For example, if one fragment in 100 thousand belongs to the pathogen and 1 million reads are obtained, we should expect to see 10 reads on average 1.
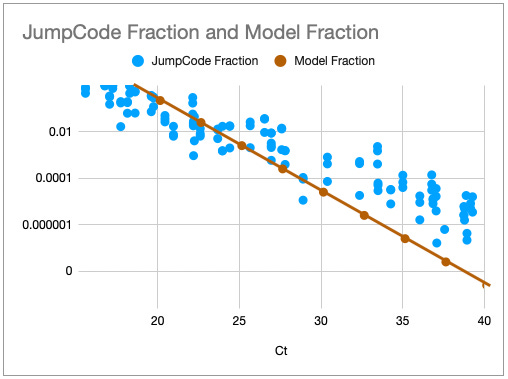
One way to think about the requirements for a MGS diagnostic is to draw a rough correspondence with the familiar Ct values from PCR for given sample characteristics. As a heuristic, Ct values of 25 represent high viral load and values over 35 are of disputed clinical relevance [46]. The relationship between viral load, Ct values and fraction of reads have been by a number of empirical studies for SARS-CoV-2 [43,47]. [table:target_reads] shows the relationship between the real fraction of target nucleic acids in a sample, measured Ct values and expected target reads based on data obtained from [43].
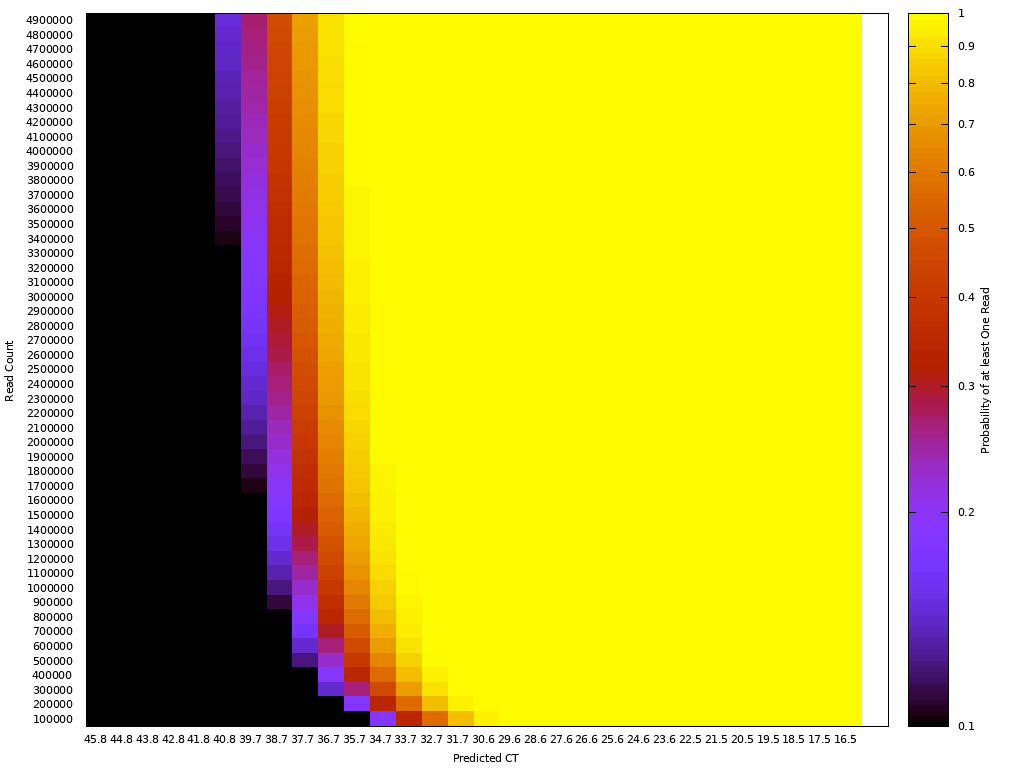
Based on this, we can map quite directly from the desired limit of detection to the required sequencing depth. For example, we see that for respiratory samples with 10 ng total nucleic acid yield, a MGS device that delivers 10M reads in the allocated runtime yields more than 10 expected reads for Ct 35. Devices capable of 1 million reads will be unreliable at these levels but may find clinical use in contexts where an order of magnitude higher limit of detection is acceptable. Given the time budget of 1 hour, this requirement leads quite directly to a throughput, which for the majority of respiratory sample applications we expect to range from 1M to 10M reads/hour. This conclusion notably does not generalize for sample types with greater abundance of human RNA such as blood, where low-abundance pathogens such as HIV are present at concentrations requiring an order of magnitude or two greater sequencing depth [48].
It is worth noting that these requirements could potentially be dramatically reduced by depleting host nucleic acids. Depletion methods remove known non-target nucleic acids (e.g., host material) from a sequencing library and doing so can reduce read depth requirements and increase detection sensitivity for pathogen nucleic acid [43]. When sequencing RNA libraries, highly repetitive ribosomal RNA (rRNA) can constitute 60-95% of a sample, making it a prime target for depletion. As indicated by interviews with practitioners, Qiagen’s FastSelect [49] is currently the most viable alternative, removing >95% rRNA (host and bacterial) in 14 minutes. There are yet to be sufficient studies examining the effectiveness of FastSelect across various sample types, but in a case where FastSelect was applied to CSF samples, practitioners report a 10x reduction in required read depth requirements (from 10-20M to 1-2M).
Unfortunately, FastSelect costs >$50/sample [49], although labs have reported [50] similar depletion results after diluting the reagent tenfold to reduce cost. The cost and time budget may be justified in many or all cases and cost reduction for rapid depletion kits are a high priority for development. However, here we conservatively assume no depletion when further building on these sequencing depth requirements.
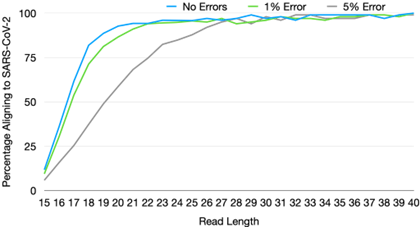
Sequencers also vary widely on two other variables: read length and single-base accuracy. Both of these features are highly desirable in research contexts where changes of even a single mutation are often the object of study. However, our simulations [51] suggest that for the task of correctly classifying a known virus, or detecting a high abundance of unknown material, read length and accuracy requirements are comparatively modest. In addition, decrease in one can be compensated by an increase in the other.
For example, for read length, we estimate that a read length of only 25 bp and single-base accuracy of 95% are sufficient for unique pathogen detection, given sufficient sequencing depth [51]. In terms of single-base accuracy, highly sensitive detection of emerging pathogens has been demonstrated in the field with error rates as high as 20-30% with early versions of long-read nanopore sequencing [52]. The fact that requirements for pathogen detection diverge so significantly from that for research applications has key implications for development directions, the subject of the next section.
.
Having determined these targets for sequencing platforms, we can now formulate concrete development goals for the previously mentioned candidate single-molecule platforms, focusing on nanopore and single-molecule optical (SMO) sequencing. While these two platform classes have a different profile of upsides and downsides for MGS, they share a key advantage in minimal time required for library preparation. ONT’s rapid sequencing kit [53] requires less than 10 minutes, and some SMO platforms do not require any library preparation after lysis and nucleic acid extraction [29,54].
It is worth emphasizing that while we expect viable solutions in the next 5-10 years to come from these two categories, we cannot confidently rule out that a viable solution will emerge from a colony-based method (e.g., if cluster generation time is reduced by an order of magnitude) or from unexpectedly fast progress on a novel technology (such as solid-state nanopores).
In nanopore sequencing, nucleic acid bases are read sequentially as they pass through protein pores embedded within a membrane. The throughput of this technology is therefore determined by the speed at which bases translocate through the pore, and the number of active pores working in parallel. Unfortunately, speed of translocation is already close to the upper bound: the translocation needs to be slowed down by a specialized motor protein in order for the electrical signal to be interpretable [55].
In a research context, the primary advantage of nanopore sequencing over other platforms lies in its long reads. Average read lengths in a standard protocol exceed 1,000 bp and the longest recorded reads exceed a million base pairs [56]. In the MGS context, long reads provide limited utility and, for nanopore sequencing in particular, present an active hindrance, as longer fragments occupy pores for a longer time period. There is therefore a direct trade-off between read length and number of reads: to a first approximation, the number of fragments sequenced scales linearly with the inverse of average read length.
A logical conclusion is that for MGS, sample preparation should include a fragmentation step to reduce the average fragment size and thus increase the chance of detecting a low-abundance pathogen. ONT MinION has 512 active channels working in parallel [57]. With sufficient saturation and the standard translocation speed of approximately 400 bp/s, we need an average fragment length of some 700 bp to get 1M reads in an hour of sequencing, and 70 bp to achieve 10M reads. The cheapest device, Flongle, has approximately 100 active channels, implying some 2 million reads in 60 minutes of sequencing.
This implies that for a sufficiently fragmented sample, nanopore platforms already meet the key requirements of fast time-to-answer, sufficient throughput, and relatively simple library preparation. However, at present, the greatest barrier for a nanopore-based diagnostic platform is the high COGS of consumables. This cost, in turn, is primarily driven by the cost of manufacturing nanopore arrays, whose cost scales linearly with chip area [38]. It follows that the two available levers for cost-optimization are reducing the required area or using lower-cost materials or fabrication procedures.
From published patents [58], it is possible to infer that the design employed by ONT presently requires the use of non-standard fabrication facilities able to process novel materials, such as MEMS fabrication facilities. Thus, the cost is likely to be driven driven by the limited capacity of MEMS fabrication facilities globally, rather than by the fundamental cost of materials and fabrication procedures.
The potential to replace MEMS fabrication with a less costly process is worth investigating as a pathway of achieving a device that fits the point-of-care specifications. There are potentially many approaches to try in parallel.
While silicon substrate arrays appear to be the most viable strategy for scaling the number of pores into the thousands or tens of thousands. For applications where a lower pore count is sufficient—such as the 126 in the Flongle—alternative methodologies may offer advantages. One such alternative is the direct fabrication of silver electrodes onto printed circuit boards, a technique successfully demonstrated on a smaller scale by the companies Nanion and Elements [59]. While there is technical uncertainty as to whether this approach can scale beyond 16 channels, scaling to some 100 channels with this approach might be feasible.
While it is difficult to predict which approach will yield the best results, we should expect that a ground-up reimplentation that aims for cost-optimization should achieve a much lower cost point. Although it is unclear whether any of these will be perceived as attractive by ONT, a number of companies, including Qitan, Genia or Nanion have entered the market and this trend is poised to continue.
Single-molecule optical technologies were brought to the market by Helicos [60] (now defunct) and PacBio [61]. Considering this class of technologies as a candidate platform may be surprising, as they have generally been embodied as costly, "fridge-sized" instruments with long library preparation (>3 hours). However, this is driven not by the fundamental needs of the platform, but rather by the market demand for high single-base accuracy and long reads. If these requirements are relaxed, SMO approaches can achieve very simple sample preparation [29] and a low cost of consumables [54].
Commonly employed library preparation techniques take many hours and are not a good fit for point-of-care MGS. However, the SMRTBells library preparation kit [62] was introduced to drive accuracy to >99.9% and read length to 15 kb, neither of which is required for MGS. Without these requirements, library preparation for optical methods can be as fast as for nanopore sequencing ( 5 minutes) and still achieve accuracies upwards of 80% combined with a compensating read length of hundreds of bases. Similarly, approaches derived from the Helicos technology have been demonstrated to work with minimal library preparation with error rates below 5% and read lengths exceeding 25 bp, making them a potential candidate for a MGS device [54].
In terms of throughput, the sequencing step alone can be achieved in a fraction of the time required for nanopore sequencing, as SMO approaches can use many more sensors working in parallel. In PacBio sequencing, imaging more than 1 million fragments in parallel is common [30] and, with a speed of 10 nucleotides per second, the run can be finished in less than a minute for the read lengths required for MGS. Another key advantage of an optical approach relative to nanopore sequencing is that it is compatible with a less complex chip and presents a clearer path to a consumable cost of $10 or less [54].
A key challenge to address will be the optimization of device cost, currently exceeding $500,000 in the case of PacBio instruments. However, achieving a cost lower than $10,000 appears feasible if instrumentation is reimagined from the ground up. For example, photonic chips need not be monolithically integrated. Consumer-grade cameras available for $300 have resolution sufficient for the accuracy requirements of an MGS diagnostic. Throughput in a low-cost platform will be limited by compatibility with low-cost cameras, but the goal of 1-10 million sensing regions (and hence reads) appears achievable.
A particularly promising emerging approach is that of electro-optical zero-mode waveguides [63], which may enable substantially lower input requirements, potentially obviating the need for random amplification of nucleic acids in a sample.
Based on the previous analysis, there are at least two independent pathways that are likely to meet the criteria for a viable point-of-care MGS device. Despite the large large amounts of private investments flowing into sequencing technology as a whole, there is a number reasons to believe that directed development can yield significant improvements over business-as-usual. A number of observations, informed largely by the authors’ experience with the sequencing landscape, as well as conversations with experts, push in this direction:
Development is driven by customer demand. The primary market for sequencing instruments is in research and high-end diagnostics (e.g. cancer). In these contexts, the primary consideration is often extraordinarily high accuracy, as the correct identification of every single base is potentially informative. The task of correctly classifying a pathogen of interest or detecting an anomaly does not require such high single-base accuracies, as we justify later. Similarly, the requirements for read length, as well as sequencing depth, are much lower.
Time to answer of hours or days is not a limiting factor in many research contexts. Typical workflows are structured around large runs, often counted in thousands of billions of bases (e.g. ONT PromethION, Illumina NovaSeq). Researchers typically care about a low cost per base for these large runs. Large, high-end instruments can minimize this cost per base.
The sequencing market is still comparatively small, dominated by a few players with relatively enforceable intellectual property, and freedom-to-operate is thus relatively limited.
Relative to the present sequencing device applications, the infectious disease diagnostic market is likely to present lower margins and lower barriers to entry. While the market is potentially large in volume, there is presently no clear demand signal that would justify the relatively large investments (at least tens of millions of dollars for a working product) necessary.
| Platform | Estimated Instruments | Runs/Week | Total Runs/Year | Run Yield (Tb) | Tb/year |
|---|---|---|---|---|---|
| ONT MinION | 5501 | 3 | 858156 | 0.05 | 42907.8 |
| ONT GridION | 782 | 3 | 121992 | 0.25 | 30498 |
| ONT PromethION 48 | 67 | 2 | 6968 | 14 | 97552 |
| Ilm Novaseq 6000 | 1485 | 3 | 231660 | 6 | 1389960 |
| Ilm NextSeq | 5430 | 3 | 847080 | 0.36 | 304948.8 |
| Ilm Miseq/Mini/iSeq | 12340 | 3 | 1925040 | 0.015 | 28875.6 |
| Ion Torrent | 2220 | 14 | 1616160 | 0.05 | 80808 |
| PacBio | 577 | 5 | 150020 | 0.03 | 4500.6 |
| MGI - Mid/Low | 2000 | 3 | 312000 | 0.72 | 224640 |
| MGI -T7 | 10 | 5 | 2600 | 6 | 15600 |
| Total | 30412 | 6071676 | 2220290.8 |
By our estimates, more than half of global sequencing capacity in terms of the number of bases sequenced annually is accounted for by Illumina NovaSeq alone [64]. NovaSeq is a $1 million instrument that yields up to 6 Tb of data per run with an error rate on the order of 0.1%.A single run can cost more than $5,000. This large share of sequencing capacity is accounted for by only some 1500 instruments in large laboratories [64].
Another key player, ONT, is known for its relatively affordable, miniaturized devices such as the MinION or Flongle. In 2021, however, fully 55.7% of ONT’s revenue was generated by its 67 PromethION [65] instruments [66], each of which can generate up to 12 Tb of data, with device costs ranging from $225,000 to $450,000.
What, then, could shift these market dynamics? Broadly, we see two approaches to this problem:
Push mechanisms have historically been successful in stimulating R&D in sequencing. The Human Genome Project and its successors (e.g. the $1,000 Genome Project) paved the way for technology development by direct public investment into R&D. Similarly, a $10 MGS device provides a challenging but achievable goal around which activity can be catalyzed by direct grants or other forms of support.
Pull mechanisms such as advanced market commitments [67] may be appropriate here, as the target product profile is readily definable in this context, and achievable in a timeframe of less than 5 years. At present, building a fab from scratch, which could pave the way towards low-cost nanopore devices, is an endeavor in the hundreds of millions of dollars and is difficult to justify given the lack of a credible demand signal. However, at volumes comparable to qPCR diagnostics (>1M units a month), such investment could be justified. Tools such as advanced market commitments, as well as credible signals of interest in an MGS diagnostic platform from national and international organizations, could pave the way towards such investments.
Widespread adoption of metagenomic sequencing for infectious disease diagnosis promises both continuous public health benefits and a greatly enhanced capacity to detect and contain future pandemics. While currently available technologies are not well suited for such widespread adoption, at least two classes of technologies, single-molecule optical and nanopore sequencing, have a good chance of achieving a combination of cost and performance that would make them a viable solution for clinicians worldwide.
This project was funded by Open Philanthropy, whose support we are grateful for. We are grateful to Rahul Arora, Jake Pencharz and Janvi Ahuja for help on the project and to Adam Marblestone for helpful comments. We also thank the researchers and clinicians who shared invaluable details on practical workflows and real-world implementation.